Среди группы наследственных болезней есть два заболевания, относящихся к самым частым причинам интеллектуальной недостаточности. Наиболее распространённая патология – синдром Дауна, связанный с наличием лишней 21-ой хромосомы в геноме человека. В этой статье мы расскажем о втором по распространенности наследственном синдроме, которое приводит к умственной отсталости, а также может сопровождаться другими клиническими проявлениями.
Заболевание получило название по фамилиям двух ученых – Джеймса Мартина и Джулии Белл – впервые описавших случай наследственной умственной отсталости у 11 мужчин из одной семьи в 1943 году. Матери при этом были интеллектуально нормальными. Генетическую причину болезни установил генетик Г. Лабс в 1969 году.
Специфика заболевания
Синдром ломкой X-хромосомы или синдром Мартина-Белл является результатом мутаций в гене FMR1 (fragile X mental retardation-1), который играет важную роль в появлении и развитии нервных связей, обучении и запоминании. Частота заболевания среди мальчиков составляет 1:4000.
Так называемая «ломкость» X-хромосомы проявляется в том, что она выглядит нетипично при специальном окрашивании, как будто один кусок отделился, хотя физически она остается цельной. Генетическая основа этого явления заключается в увеличении числа тринуклеотидных повторов CGG в гене FMR1.
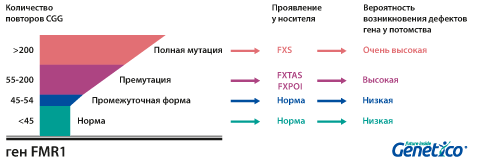
У здоровых людей число повторов в этом гене колеблется от 5 до 54. Если их больше 200, то наработка белка с гена FMR1 нарушается, что приводит к развитию синдрома Мартина-Белл и клиническому проявлению заболевания. Премутационное состояние — это количество повторов CGG от 55 до 200. В таком случае заболевание у людей в типичной форме не проявляется, но чем больше повторов в этом гене у носителя, тем выше вероятность того, что у потомков количество повторов будет больше 200 и возникнет заболевание. В случае носительства премутации при формировании половых клеток количество повторов может увеличиваться, поэтому если у родителя их число от 55 до 200, то высока вероятность рождения ребенка с полной мутацией в гене FMR1 и синдромом Мартина-Белл. При этом носительство премутационного состояния будущих родителей неравнозначно по вероятности возникновения мутантного аллеля у их детей: если носитель – мама, то вероятность значительного увеличения числа повторов выше. Количество повторов от 45 до 54 является промежуточной формой, которая не имеет никакого влияния на здоровье человека, но может приводить к проблемам у будущих поколений, как и в случае премутационного состояния гена.
Важно учитывать, что наследование и развитие заболевания зависит от пола, так как ген FMR1 находится на Х-хромосоме. У мужчин только одна Х-хромосома, которую они получают от матери. Поэтому, в случае если она оказалась «ломкой», у них проявляется заболевание. У женщин две Х-хромосомы, однако активно работает только одна из них, вторая «выключается» или инактивируется. Поэтому наличие одной Х-хромосомы с полной мутацией в гене FMR1 может не проявляться клинически, в случае инактивации именно «ломкой» хромосомы, или приводить к развитию заболевания в 30-50% случаев. Мужчина с ломкой Х-хромосомой передает её всем дочерям, но ни одному из сыновей. Женщина с мутантной хромосомой имеет шансы передать её как сыновьям, так и дочерям с равной вероятностью.
Премутационное состояние гена влияет как на судьбу потомков носителя, так и непосредственно на его здоровье:
Развитие первичной недостаточности яичников (FXPOI) (снижение овариального резерва и наступление менопаузы до 40 лет). Мутация FMR1 является причиной преждевременного истощения яичников у 5% женщин с этим диагнозом. Среди носительниц премутации примерно у четверти развивается это состояние. Оно влияет не только на общие репродуктивные возможности, но и на подбор протокола стимуляции при ВРТ, так как часто оказывается причиной бедного ответа яичников на стимуляцию. Интересно, что по данным, полученным в центре Genetico, хотя этот фактор влияет на число получаемых в цикле эмбрионов, он не приводит к увеличению доли анеуплоидных эмбрионов.
Тремор/атаксия, ассоциированные с ломкой Х-хромосомой (FXTAS). Это состояние чаще развивается у мужчин: при носительстве премутации мужчиной проявляется в 33% случаев, а при носительстве премутации женщиной – лишь в 5-10%. Синдром FXTAS начинает проявляться в пожилом возрасте. Наблюдается тремор, шаткая походка, может страдать речь.
Диагностика синдрома Мартина-Белл
Метод диагностики, используемый в лаборатории Genetico, основан на использовании полимеразной цепной реакции с особым набором праймеров, позволяющих не только детектировать нормальное, премутационное и мутационное состояния, но и точно определить количество повторов в случаях, когда их меньше 200. Такое исследование позволяет выявить синдром ломкой X-хромосомы на молекулярном уровне, а также оценить вероятность рождения больного ребенка и возможность развития у пациента расстройств, связанных с увеличенным количеством повторов в гене FMR1. Метод также позволяет детектировать наличие AGG повторов среди повторов CGG. Полагают, что участки AGG, прерывающие длинную последовательность из CGG повторов, придают ДНК устойчивость и снижают риск увеличения количества повторов в следующем поколении.
Генетический тест, определяющий количество повторов в гене FMR1, рекомендуется пройти в первую очередь женщинам с синдромом преждевременного истощения яичников или с выявленной неслучайной инактивацией Х-хромосомы (косвенный признак), семьям, в которых есть сыновья с интеллектуальной недостаточностью. Также анализ состояния гена FMR1 необходим:
1) женщинам с репродуктивными проблемами или нарушениями фертильности, связанными с повышенным уровнем фолликулостимулирующего гормона (ФСГ);
2) пациентам с интеллектуальной недостаточностью и их родственникам;
3) тем, у кого в семье были случаи синдрома ломкой Х-хромосомы или умственной отсталости без точного диагноза;
4) женщинам, у родственников которых наблюдались нарушения, связанные с премутационным состоянием в гене FMR1;
5) пациентам с поздно проявившимся тремором и мозжечковой атаксией (нарушения согласованности работы мышц из-за поражения систем мозга, управляющих движением мышц).
Результаты исследования
В случае обнаружения бессимптомного носительства полной мутации в гене FMR1 у женщины может быть рекомендовано использование донорских ооцитов или проведение преимплантационной генетической диагностики (ПГД) с целью исключить возможность проявления синдрома у ребенка. Также важно правильно оценивать риск рождения больного ребенка в случае премутационного состояния гена FMR1 у будущих родителей. В таком случае по результатам теста рекомендуется консультация врача-генетика.
Автор: Очир Мигяев
Стажер лаборатории Genetico
Источники:
Hagerman R.J. Fragile X Syndrome: Diagnosis, Treatment and Research. JHU Press, 2002.
Van der Molen M.J.W., Huizinga M., Huizenga H.M., Ridderinkhof K.R., Van der Molen M.W., Hamel B.J.C., Curfs L.M.G., Ramakers G.J.A. Profiling Fragile X Syndrome in males: Strengths and weaknesses in cognitive abilities // Research in Developmental Disabilities, 2010.
Merenstein S.A., Shyu V., Sobesky W.E., Staley L., Berry-Kravis E., Nelson D.L. et al. Fragile X syndrome in a normal IQ male with learning and emotional problems. J. Am. Acad. Child Adolesc. Psychiatry, 1994.
Sitzmann, A., Hagelstrom, R., Tassone, F. et al. Rare FMR1 gene mutations causing fragile X syndrome: A review. American journal of medical genetics. Part A, 2018.
Туйчибаева, Н., Ф. Алимходжаева, К. Губайдулина, Б. Ганиев, и И. Расулова. «КЛИНИКО-ГЕНЕТИЧЕСКИЕ И ДИФФЕРЕНЦИАЛЬНО ДИАГНОСТИЧЕСКИЕ КРИТЕРИИ СИНДРОМА МАРТИНА-БЕЛЛА». Медицина и инновации, т. 1, вып. 4, январь 2022.
Также, вам может быть интересно
Хромосомные болезни
Почти каждая клетка человеческого организма содержит полный диплоидный геном. Это геном с двойным, или парным набором хромосом, который наследуется от обоих родителей. Диплоидный геном состоит из ДНК, организованной в 46 хромосом: 22 схожих аутосомных пары, а также половые хромосомы XX у женщин и XY — у мужчин.
Большинство клеток в организме обычно содержат 46 хромосом, организованных в 23 пары. Числовое обозначение имеют 22 хромосомы (1–22). А последняя, 23-я пара хромосом определяет пол человека и имеет буквенное обозначение — XY у мужчин, и XX у женщин.
Гипертриглицеридемия – это патологическое состояние, при котором в крови повышается уровень триглицеридов. В организм они могут поступать двумя путями – синтезироваться в печени и попадать вместе с пищей. Соответственно и причины повышения их уровня могут быть разными.
При наследственной форме заболевания онкоклетки размножаются под слизистой оболочкой желудка. Это приводит к развитию «диффузной» онкологии, а не к появлению солидной опухоли. Это также увеличивает вероятность того, что рак распространится на другие органы и ткани, включая печень и кости.